
花季少女肚大如鼓、2岁男孩指甲莫名脱落……这些罕见病你了解吗?
文章来源:admin 时间:2024-03-20
每年2月的最后一天是国际罕见病日。每年这个时候,我们都会讲述罕见病患者和医护人员相互信任,共同去寻找希望、寻找自我的故事。因为我们知道,百万分之一、千万分之一的发病率,对每一个患者每一个家庭就是百分之一百。
随着国家和社会各界的关注重视、诊疗技术水平和用药保障水平的不断提升,越来越多的罕见病患者得到救治、帮助。
今年是第17个国际罕见病日,医院官方微信继续推出主题为“‘罕’卫不凡”的系列报道,讲述每一个百分百的生命故事。
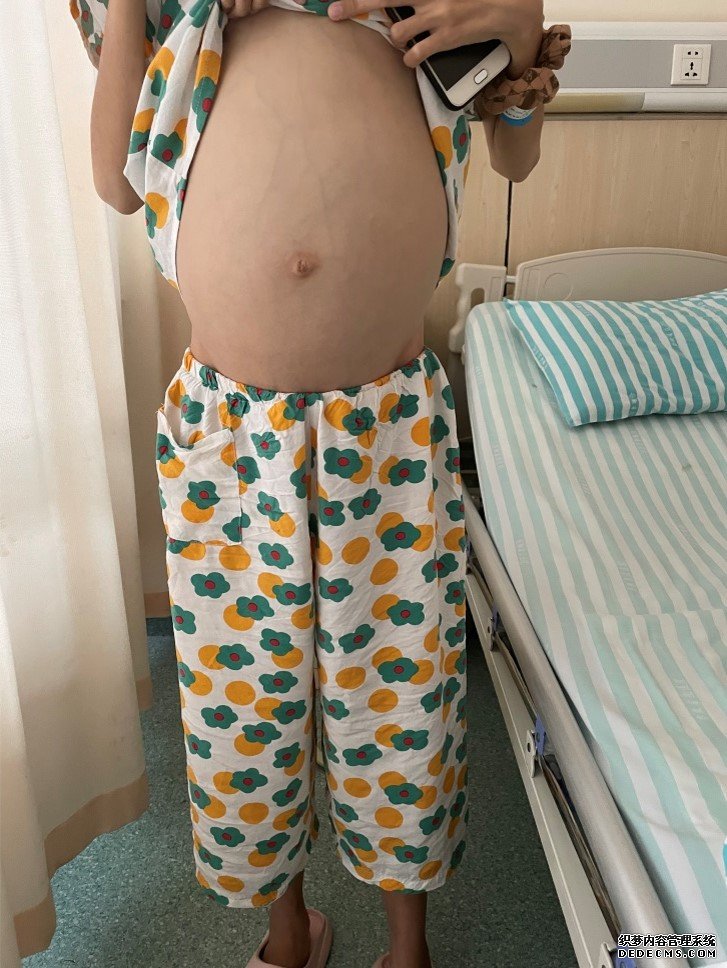
2016年,在小惠13岁的时候,妈妈发现她的肚子越来越鼓,身材也十分瘦小,走起路来就像一只小企鹅。妈妈带小惠到医院检查后发现,她的脾脏增大,而且贫血伴有血小板减少。医生为小惠做了骨髓穿刺检查,发现她的骨髓里有长得像洋葱皮一样的大细胞,叫“戈谢细胞”。
医生又询问了小惠的家庭情况,得知她的父母是近亲结婚,小惠的妹妹也有脾脏肿大症状。根据这些信息,医生进一步为小惠完善了相关检查,发现她的葡萄糖脑苷脂酶活性降低。综合以上信息,小惠最终确诊为戈谢病(GaucherDisease,GD),基因检测在小惠妈妈身上找到了同样的致病基因。
但在当时,治疗戈谢病的特效药非常昂贵,一支就要2万余元,算起来每年治疗的花费近100万元,小惠的家庭实在无法承受,她的治疗也就被搁置了。
随着小惠年龄的增长,她的肚子越来越大。到了19岁的时候,她的肚子已经胀得影响吃饭了,肚皮上的青色血管根根分明,面色苍白,还经常流鼻血,月经也突然不来了……于是家人带着小惠来到了中国科大附一院(安徽省立医院)就诊,这一次血液内科的医生给小惠和她的兄弟姐妹们做了全面细致的检查。
血液内科副主任医师皖湘介绍,戈谢病是一种罕见的常染色体隐性遗传性代谢病,是由于机体葡萄糖脑苷脂酶的活性缺乏造成体内葡萄糖脑苷脂在肝、脾、肾、骨骼、肺、甚至脑的巨噬细胞中贮积,形成典型的“戈谢细胞”,导致组织器官出现病变和功能损害。患者表现为不明原因的肝脾肿大、贫血、血小板减少、骨痛、神经系统症状等。因为患者肚子肿胀,身材矮小,也被称为“企鹅宝宝”。在我国,平均每20万-50万人中可能出现一例。
目前,戈谢病的特效治疗药已经纳入了国家罕见病医保报销范围,综合报销比例最高能达到95%,相关的检测和筛查也可以免费进行。在了解到相关政策后,原本打算做手术切掉巨大脾脏的小惠选择定期注射特效药进行治疗。经过半年的规范治疗,小惠的脾脏已经较之前缩小了1/3,血象也得到了恢复,身体长高也长胖了。此外,小惠的妹妹经过免费的疾病筛查也确诊为戈谢病,并且接受了特效治疗药的替代治疗。
皖湘提醒,葡萄糖脑苷脂酶活性检测是戈谢病诊断的金标准,对于不明原因脾脏肿大和(或)血小板减少的患者,需要考虑到戈谢病并进行相关检测,早期确诊和治疗对于患者至关重要。有相关家族遗传病史的患者孕前要做好戈谢病遗传咨询,完善孕期检查以保障新生儿的健康。
5岁的男孩小穆,从小就有着“大头”的外号。据小穆妈妈介绍,在小穆1岁时,因为疝气反复发作就诊于当地医院,当时医生觉得小穆头围偏大,建议进一步完善检查,但是他们没有放在心上。
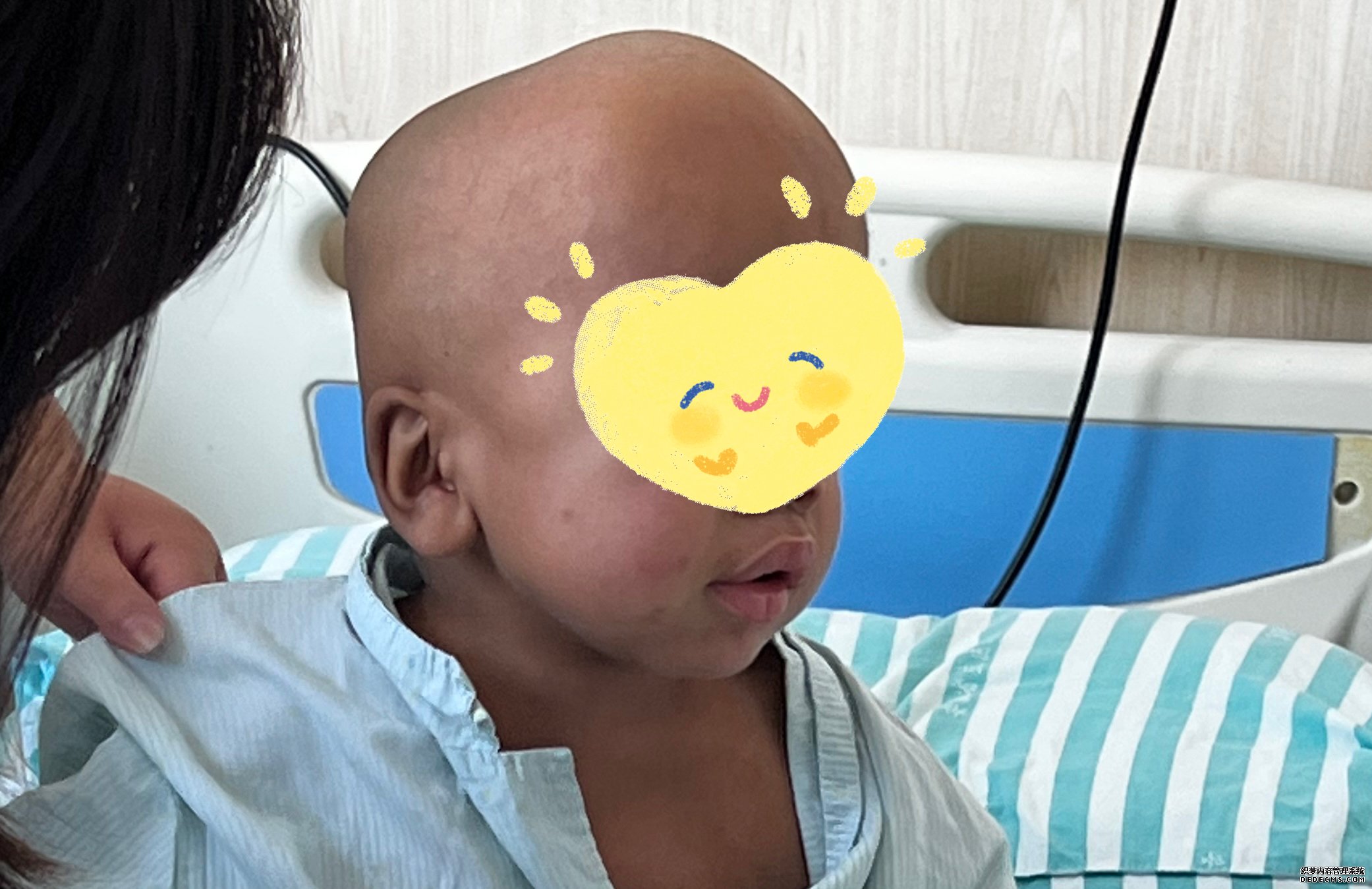
随着小穆逐渐长大,他与同龄人的差异变得越来越明显:头围与日俱增,智力发育不完全,只能单音节发音,手脚活动度差,骨骼变形。家人便带着小穆前往外地就医,医生立即为小穆进行了一系列检查,通过酶学和基因检测,明确诊断为黏多糖贮积症I型(mucopolysaccharidoses,MPS-I)。
黏多糖贮积症(mucopolysaccharidosis,MPS),又称黏多糖症,是一组罕见的遗传性代谢障碍性疾病。主要是因为机体缺乏降解黏多糖所需要的酶,导致黏多糖在体内不断沉积,最终影响到各个组织和器官的功能,引发一系列症状和并发症。因为这类患者很难生存到成年,所以也被称为“长不大”的黏宝宝。
中国科大附一院(安徽省立医院)血液内科主任朱小玉介绍,MPS在儿童时期就可以出现症状,包括身材矮小、面容特征异常、智力发育迟缓、腹部突出、呼吸功能和心功能不全、骨骼和关节畸形等。随着疾病的进展,这些症状会逐渐加重,并且还会伴随一些并发症,如听力损害、角膜混浊、肝脏功能异常等。
酶替代治疗(ERT)和异基因造血干细胞移植(allo-HSCT)是针对MPS的特异性治疗方法。多方打听后,小穆的父母决定带他到我院血液内科接受脐带血移植治疗。朱小玉指出,脐带血是胎儿娩出、脐带结扎离断后残留在胎盘和脐带中的血液,含有珍贵的造血干细胞,可用于造血干细胞移植,是新生儿带给人类的厚礼。对于患有遗传性疾病如MPS患者来说,脐带血移植是一种理想的治疗选择。
2023年1月份,经科室讨论,在符合移植相关条件的情况下,小穆成功接受了脐带血移植,移植过程顺利。移植后14个月,小穆复查各项指标正常,血IDUA基因(MPS-I型的致病基因)测序无突变,IDUA也恢复至正常水平。让人欣喜的是,先前需要妈妈牵着走路的小穆,已经能够独立行走。
在小瑞瑞2岁多的时候,右手食指的指甲突然开始萎缩脱落,身上也会莫名出现瘀斑、口腔黏膜白斑。在半年多之后的一次体检中,小瑞瑞的血常规检查结果显示,他的血小板和血红蛋白明显低于正常水平。
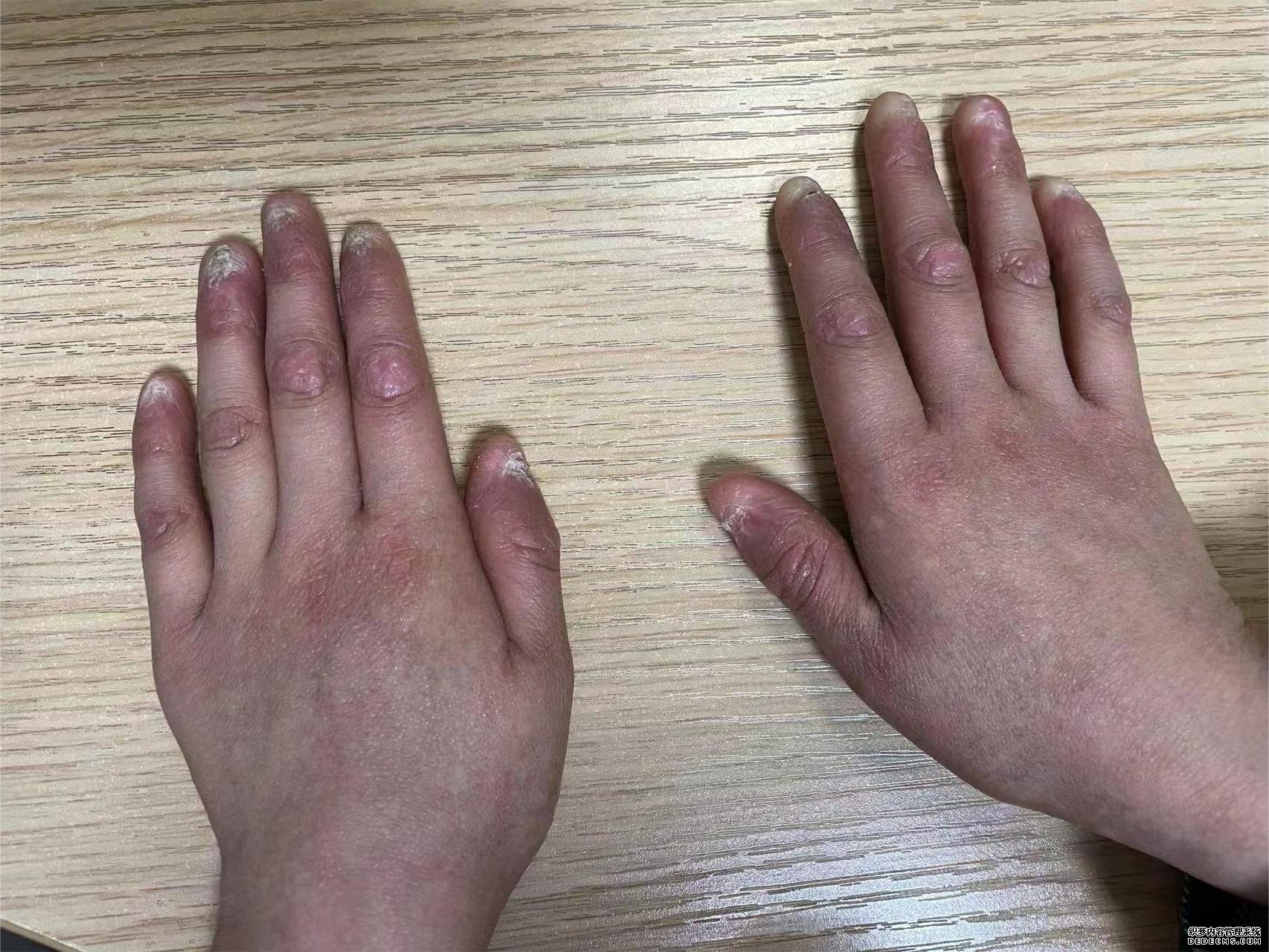
小瑞瑞的父母赶紧带他到医院就诊,医生为他完善了骨髓穿刺、基因检测等多项检查,发现他骨髓造血衰竭、TINF2基因突变,最终确诊为先天性角化不良。随着时间推移,小瑞瑞萎缩脱落的指甲越来越多,贫血和血小板减少也越来越严重,没有力气和其他小朋友一起玩耍,还经常需要住院输血治疗。
后来,小瑞瑞的父母了解到造血干细胞移植可以解决骨髓衰竭的问题,便慕名带他来到了我院血液内科就诊。
皖湘介绍,先天性角化不良(dyskeratosiscongenital,DC)是一种罕见的进行性骨髓衰竭综合征,典型临床表现为皮肤色素沉着、指(趾)甲发育不良和口腔黏膜白斑等“三联征”,发病率为1/100万,且多见于男孩。除了“三联征”以及骨髓衰竭以外,患者肺部、眼睛、胃肠道、大脑等几乎全身所有器官和组织都有可能受到影响,罹患癌症的风险也比健康人群高。目前,先天性角化不良没有特效的治疗方式,针对造血功能衰竭可以进行异基因造血干细胞移植治疗。
2019年4月,小瑞瑞在中国科大一附院血液内科进行了异基因造血干细胞移植,供者是他的同胞姐姐,HLA配型完全相合。移植后小瑞瑞的血常规逐步恢复了正常,不需要频繁地住院输血,精气神也好了很多,可以和其他小朋友一起玩耍了。在皖湘医生门诊的随访过程中,这个坚强的小朋友也一天天长大了。
中国科大附一院(安徽省立医院)血液内科联合多学科、多中心,致力于血液系统疑难病、罕见病的诊治,期望为罕见病的诊疗与管理积累经验,推动诊疗规范的制定,为患者带来更好的疗效。
近年来诊治的罕见病包括:范可尼贫血、先天性角化不良、先天性纯红细胞再生障碍性贫血、阵发性睡眠性血红蛋白尿症、湿疹血小板减少综合征、先天性粒细胞缺乏、先天性角化不良、丙酮酸激酶缺乏症、黏多糖贮积症、X连锁无丙种球蛋白血症、原发性联合免疫缺陷病、儿童极早发炎症性肠病、Castleman病、POEMS综合征等。
互联网新闻信息服务许可证:增值电信业务经营许可证:皖B2-20080023